About multimodal single-cell data
The flow of genetic information in cells
细胞是生命的基本单位。一切生物都是由细胞构成的。这包括树木、鱼类、人类、细菌、真菌等。细胞有各种形状和大小,但无论我们从怎么看,它们都有一些共同的属性。例如,所有的细胞都是 membrane-bound(细胞膜)。这意味着他们在内部和外部之间有一个明确的界限。所有的细胞都含有某种形式的细胞器,这些细胞器是执行特定功能的特殊的子结构。而且,所有的细胞都包含三层遗传信息: DNA、 RNA 和 蛋白质。

DNA定义有机体。事实上,你可以通过基因组移植(genome transplantation)来改变细菌的种类。 然而,DNA不是功能性的(functional)。它包含一套必须转化成RNA然后再转化成蛋白质的指令。在大多数情况下,你可以把RNA看作是DNA和蛋白质之间的信使。DNA是由基因组成的,这些基因包含如何制造蛋白质的指令。蛋白质在细胞中负责执行生物功能,如代谢葡萄糖为细胞创造能量。一般来说,你体内的每一种蛋白质都是由一个基因编码的。
尽管你体内的所有细胞都包含相同的基因组,相同的DNA,但这数万亿个细胞执行着非常不同的生物功能。免疫细胞、神经元或肌肉细胞之间的区别是由这些细胞内基因的开启或关闭决定的。当一个基因被激活时,就会产生更多的RNA副本,从而增加蛋白质的产量。我们直到蛋白质数量的调节同时发生在转录阶段(DNA $\rightarrow$ RNA) 和翻译阶段(RNA $\rightarrow$ 蛋白质)。
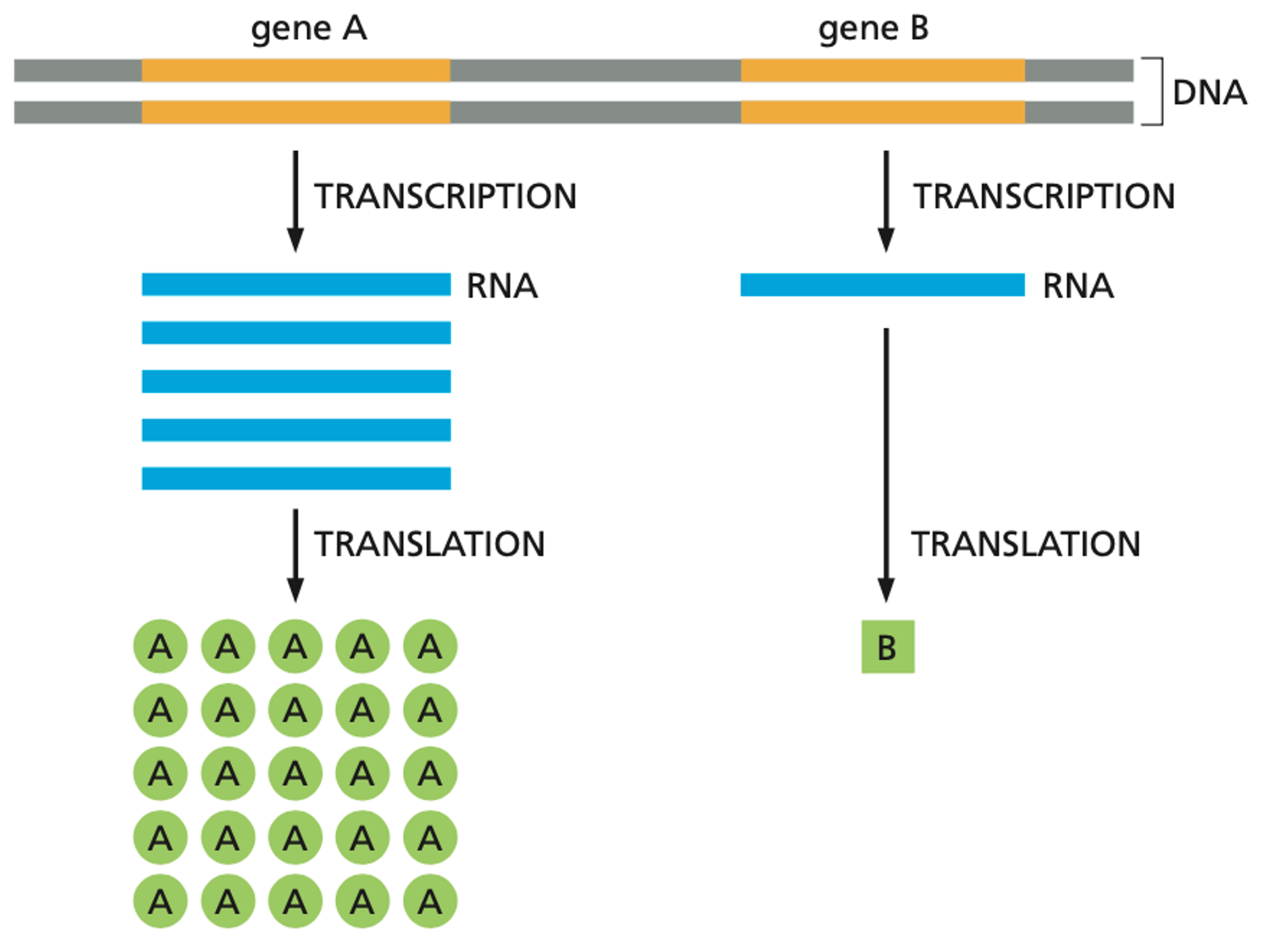
因为我们知道不同类型的细胞与不同水平的RNA和蛋白质有关,所以能够在单个细胞的水平上测量这些分子的丰富程度是非常有用的。这不仅让我们对体内不同种类的细胞有了精细的了解,还让我们深入了解了同一组DNA指令是如何在全身被如此不同地解释的。
单细胞基因信息测量的前景是,通过更好地了解这些信息在我们的细胞和组织中是如何流动的,我们可能会更好地了解在疾病的背景下哪里出了问题。
A rough history of single-cell technologies
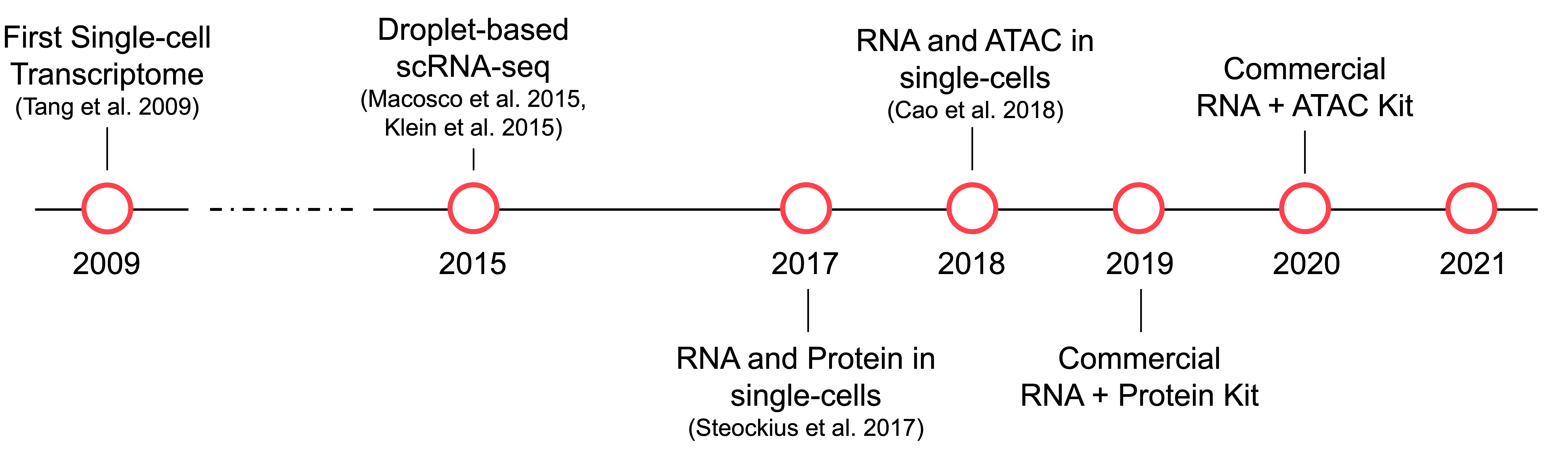
Eberwine等人(1992)首次使用分子探针对单细胞RNA进行了测量。直到2009年,Tang等人描述了单个细胞(小鼠卵裂球)的转录组序列。在接下来的6年里,一些创新被开发出来以提高单细胞RNA测序(scRNA-seq)的吞吐量。也许最具影响力的是由Klein et al. (2015) and Macosko et al. (2015)同时描述了两种 用于将单个细胞捕获到 oil emulsion 的 nanoliter droplets 中的 droplet-based 的协议。有了 droplet-based 的单细胞方法,就有可能用数万个细胞进行实验。

接下来在单细胞测量的主要创新是 Steockius et al. (2017) and Cao et al. (2018) 引入了多模态单细胞数据, 分别测量RNA和蛋白质 或 chromatin accessibility 和 RNA。 第一种方法叫做 ` Cellular Indexing of Transcriptomes 和 Epitopes by Sequencing (CITE-seq), 测量每个细胞的所有 RNA 以及 ~10-200个细胞表面markers。 第二种方法, 称作 Multiome Assay for Transposase-Accessible Chromatin using sequencing (ATAC) + Gene Expression, 提供一个通过基因组和所有基因的基因表达 测量染色体 accessibility。与 CITE-seq` 不同, 这种 Multiome technology 提供在细胞中横跨所有 DNA 和 RNA 的一种视角。第二年,Nature Methods将单细胞多模态组学评为2019年度方法,指出这些方法为先进统计和计算方法的发展打开了前所未有的机遇。
令我们兴奋的是,到2021年,我们可以使用强大的商业可用试剂来创建参考基准数据集,以推动多模态单细胞数据集成的创新。
Measuring multiple modalities in single cells
我们知道 DNA 必须是 accessible 的(ATAC data) 以生产 mRNA (expression data), 以及 mRNA反过来作为一种模板生产蛋白质(protein abundance)。 这些过程通常是由它们产生的相同分子控制的:例如,一种蛋白质可以结合DNA来阻止更多mRNA的产生。理解这些调控过程对于生物合成和药物发现是革命性的。任何从一种模态预测另一种模态的方法可以解释这些调控过程, 而且对于多模数据的需求表明这是重要的。
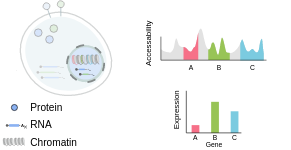
它还能够捕获同一细胞中的RNA和蛋白质表达。细胞表面的蛋白质不仅对识别不同的细胞群很重要,而且这些蛋白质也有功能作用,特别是在免疫系统中。目前,我们只能测量单个细胞上大约100-200种蛋白质,但我们知道细胞含有数万种(或更多)独特的蛋白质。然而,这一信息对于解开 转录相似但表达不同功能的表面 markers的 细胞群的身份至关重要。
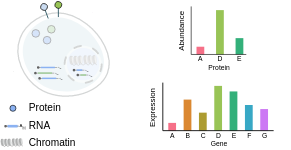
Multimodal data as a basis for benchmarking
开发生物系统的机器学习方法由于难以获得标签而变得复杂。通常,机器学习任务依赖于手动标注(如图像或自然语言查询)、动态测量(如纵向健康记录或天气)或多模态测量(如翻译或文本到语音)。然而,在单细胞生物学的背景下,这就更加复杂了。
单细胞数据, 标注是不可行的。 数据是有噪声的并且没有完全理解细胞类型快速进化的描述。 同样,由于目前的测量技术涉及到破坏细胞,所以无法纵向测量细胞中的所有RNA。然而,有了多模态单细胞数据,我们现在可以直接观察到同一细胞中的两层遗传信息。这提供了一个机会,利用这两组数据在同一细胞中同时出现的事实作为标签。这类似于用两种语言表达相同的情感,为机器翻译提供了标签。
然而,由于这些技术相对较新,大多数公开可用的数据集都是为探索而设计的,而不是 benchmarking。为了建立multimodal single-cell data integration的竞赛,我们开始创建一个适合目标的benchmarking数据集。
The benchmarking dataset
我们目前正在为竞赛创建一个基准数据集。将有两种类型的数据:
- 使用 ` Biolegend TotalSeq™-B Universal Cocktail v1.0
的10X Genomics Single Cell Gene Expression with Feature Barcoding` 测量的单细胞 RNA 和 蛋白质的 Joint profiling。 - 使用
10X Genomics Single Cell Multiome ATAC + Gene Expression Kit单核 RNA 和 染色体 accessibility 的 Joint profiling。
Download the data
数据的当前形式在S3上是公开的。要下载数据,首先在您的计算机上安装AWS CLI: https://aws.amazon.com/cli/。
您可以使用以下命令将数据下载到本地计算机(数据集大小大约有1.2GiB):
1
aws s3 sync s3://openproblems-bio/public/explore /tmp/public/ --no-sign-request
您将找到以下文件:
1
2
3
4
5
6
7
explore
├── LICENSE.txt
├── README.txt
├── cite/cite_adt_processed_training.h5ad
├── cite/cite_gex_processed_training.h5ad
├── multiome/multiome_atac_processed_training.h5ad
└── multiome/multiome_gex_processed_training.h5ad
这些都是AnnData h5ad文件,如下节所述。
Data file format
训练数据可以在 ` AnnData h5ad 文件中得到。 更多关于 AnnData 的信息可以在 [这里](https://openproblems.bio/neurips_docs/submission/quickstart/) 找到。 你可以使用 AnnData.read_h5ad()` 函数下载这些文件。 最简单的开始方式是 在 Saturn Cloud 上注册一个免费的 Jupyter 服务器。
1
2
3
4
5
!pip install anndata
import anndata as ad
adata_gex = ad.read_h5ad("cite/cite_gex_processed_training.h5ad")
adata_adt = ad.read_h5ad("cite/cite_adt_processed_training.h5ad")
您可以在我们的数据中找到用于探索数据的代码示例 exploration notebooks。
Preprocessing
为了便于探索数据,每个数据集都经过预处理,以去除低质量的细胞和重复数据。下面几节详细介绍每种数据模态的细节。
Preprocessing of gene expression (GEX)
在这个数据集中,使用如 10X Multiome Product Guide 描述的 3 种 细胞核RNA 捕获测量方法得到 基因表达。 注意,并非所有的RNA都是在细胞核中发现的。细胞核RNA 和 细胞质 RNA 以及 单核RNA 和 单细胞RNA序列 的比较之前已经有工作报导(e.g. Bakken 2018; Abdelmoez 2018, Lake 2017)。
对于基因表达数据, 根据线粒体内容过滤细胞, UMI每个细胞计数, 以及每个细胞检测到的基因。 使用 scran 计算 Size factors 并保存在 ` adata.obs[“size_factors”]`。
然后通过UMI计数除以 size factors 将每个细胞的计数标准化。原始计数存储在 adata.layers["counts"]。 size factor 规范化后的计数存储在 adata.X。
最后, 规范化后的计数进行 log1p transformed。规范化化后的计数存储在 adata.layers["log_norm"]。
在 这里 可以找到关于 single-cell analysis 最佳实践的更多信息。
Preprocessing of ATAC
蛋白质数据使用 134个细胞表面标记和6个 isotype controls 的 TotalSeq™-B Human Universal Cocktail, V1.0 测量得到。 isotype controls 存储在 adata.obsm["isotype_controls"]。 这些对照不针对任何人类蛋白,其表达应考虑背景。
ADT蛋白的测定是基于ADT总数进行质量控制的(样本范围从1100-1200到24000), 每个细胞中捕获的蛋白质数量(下限为80)和6个 isotype controls 的ADT计数相加(范围从1到100)。
由于捕获的ADT总数是有限的,如果存在高度丰富的蛋白质,绝对ADT计数似乎较低。为了解释这一影响,使用中心对数比(CLR)转换进行归一化。CLR计数存储在 adata.X 并且原始计数存储在 adata.layers["counts"]。
Metadata
关于特征更多的信息可以在每个对象的 .var 和 .obsDataFrame 中找到。
Gene expression observation metadata
GEX adata 对象有以下的列:
.obs.index:每个批量观测标签添加的细胞条码 。.obs["n_genes_by_counts"]: 一个细胞中至少有一个计数的基因的数量。.obs["pct_counts_mt"]: 对应线粒体基因的 UMI 计数的百分比。.obs["n_counts"]: cell中检测到的UMIs数量。.obs["n_genes"]: 细胞中检测到的基因数量。.obs["size_factors"]: 细胞估计的 size factor。 见 OSCA Ch. 7 - Normalization。.obs["phase"]: 由 scanpy.tl.score_genes_cell_cycle 计算得到的每个细胞的cell cycle phase。.obs["leiden_final"]:.obs["atac_ann"]: 来自joint ATAC数据的细胞的细胞类型注释。.obs["cell_type"]: 来自 GEX 数据的细胞的细胞类型注释。.obs["pseudotime_order_GEX"]: 对数据中发展轨迹标注的 diffusion pseudotime 标注。.obs["batch"]: 取样细胞的批次。 对于 Site1 Donor 1格式是s1d1。
Gene expression feature metadata
GEX adata.var DataFrames 有以下的列:
.var.index: 对于每个基因的 ` Ensembl Gene Names` 。.var["gene_ids"]: 用于追踪 随着时间变化基因名字变化的 基因的唯一的 ` Ensembl Gene Names`。.var["feature_types"]: 将每个特征表示为基因表达特征。 所有基因都应该为GEX。.var["genome"]: 用于读取映射的 ` Genome Assembly`。var["n_cells-[batch]"]: 在[batch]中检测到基因的细胞的数量。.var["highly_variable-[batch]"]: 基因是否在[batch]中被确定是highly variable的。
ATAC observation metadata
ATAC feature metadata
ATAC adata.var DataFrames 有如下的列:
.var.index- 对于每个 ATAC peak 的 Genomic coordinates, 它与参考基因组直接相关, 并按下列格式包括chromosome name*、起始位置和结束位置:chr1-1234570-1234870。.var["feature_types"]: 表示每个特征为一个基因表达特征。所有的 peaks 应该都是ATAC。.var["n_cells-[batch]"]: 在检测到 peak 的[batch]中, 细胞的数量。